Pyruvate kinase deficiency
PK deficiency; PKD
Pyruvate kinase deficiency is an inherited lack of the enzyme pyruvate kinase, which is used by red blood cells. Without this enzyme, red blood cells break down too easily, resulting in a low level of these cells (hemolytic anemia).
Images
Causes
Pyruvate kinase deficiency (PKD) is passed down as an autosomal recessive trait. This means that a child must receive a non-working gene from each parent to develop the disorder.
There are many different types of enzyme-related defects of the red blood cell that can cause hemolytic anemia. PKD is the second most common cause, after glucose-6-phosphate dehydrogenase (G6PD) deficiency.
PKD is found in people of all ethnic backgrounds. But, certain populations, such as the Amish, are more likely to develop the condition.
Symptoms
Symptoms of PKD include:
- Low count of healthy red blood cells (anemia)
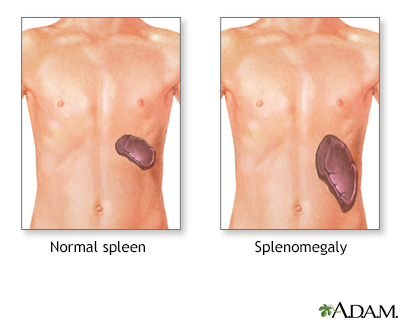
- Swelling of the spleen (splenomegaly)
- Yellow color of the skin, mucous membranes, or white part of the eyes (jaundice)
- Neurologic condition, called kernicterus, that affects the brain
- Fatigue, lethargy
- Pale skin (pallor)
- In infants, not gaining weight and growing as expected (failure to thrive)
- Gallstones, usually in the teens and older
Exams and Tests
The health care provider will perform a physical exam and ask about and check for symptoms such as an enlarged spleen. If PKD is suspected, tests that will likely be ordered include:
- Bilirubin in the blood
- CBC blood test
- Genetic testing for changes in the pyruvate kinase gene
- Haptoglobin blood test
- Osmotic fragility
- Pyruvate kinase activity
- Stool urobilinogen
Treatment
People with severe anemia may need blood transfusions. Removing the spleen (splenectomy) may help reduce the destruction of red blood cells. But, this does not help in all cases. In newborns with a dangerous level of jaundice, the provider may recommend an exchange transfusion. This procedure involves slowly removing the infant's blood and replacing it with fresh donor blood or plasma.
Someone who had a splenectomy should receive the pneumococcal vaccine at recommended intervals. They also should receive preventive antibiotics until age 5.
Support Groups
More information and support for people with PKD condition and their families can be found at:
- National Library of Medicine - MedlinePlus -- medlineplus.gov/genetics/condition/pyruvate-kinase-deficiency/
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/pyruvate-kinase-deficiency/
- NIH Genetic and Rare Diseases Information Center -- rarediseases.info.nih.gov/diseases/7514/pyruvate-kinase-deficiency
Outlook (Prognosis)
The outcome varies. Some people have few or no symptoms. Others have severe symptoms. Treatment can usually make symptoms less severe.
Possible Complications
Gallstones are a common problem. They are made of too much bilirubin, which is produced during hemolytic anemia. Severe pneumococcal disease is a possible complication after splenectomy.
When to Contact a Medical Professional
See your provider if:
- You have jaundice or anemia.
- You have a family history of this disorder and are planning to have children. Genetic counseling can help you know how likely it will be that your child would have PKD. You can also learn about tests that check for genetic disorders, such as PKD, so that you can decide if you'd like to have these tests.
Related Information
EnzymeHemolytic anemia
Autosomal recessive
Glucose-6-phosphate dehydrogenase deficiency
Anemia
References
Gallagher PG. Hemolytic anemias: red blood cell membrane and metabolic defects. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 147.
Remiker AS, Brandow AM. Enzymatic defects. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 512.
BACK TO TOPReview Date: 12/31/2023
Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.

Health Content Provider
06/01/2025
|
A.D.A.M., Inc. is accredited by URAC, for Health Content Provider (www.urac.org). URAC's accreditation program is an independent audit to verify that A.D.A.M. follows rigorous standards of quality and accountability. A.D.A.M. is among the first to achieve this important distinction for online health information and services. Learn more about A.D.A.M.'s editorial policy, editorial process and privacy policy. A.D.A.M. is also a founding member of Hi-Ethics. This site complied with the HONcode standard for trustworthy health information from 1995 to 2022, after which HON (Health On the Net, a not-for-profit organization that promoted transparent and reliable health information online) was discontinued. |
The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. © 1997- 2025 A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited.
 All rights reserved.
All rights reserved.