Amyotrophic lateral sclerosis (ALS)
Lou Gehrig disease; ALS; Upper and lower motor neuron disease; Motor neuron disease
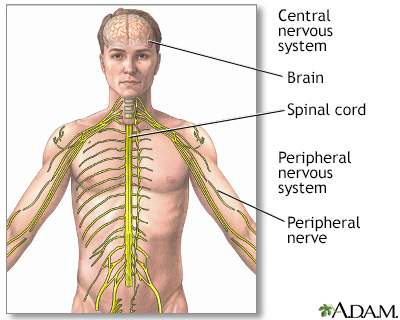
Amyotrophic lateral sclerosis, or ALS, is a disease of the nerve cells in the brain, brain stem and spinal cord that control voluntary muscle movement.
ALS is also known as Lou Gehrig disease.
Images
Causes
One in 10 cases of ALS is due to a genetic variant. The cause is unknown in most other cases.
In ALS, motor nerve cells (neurons) waste away or die, and can no longer send messages to muscles. This eventually leads to muscle weakening, twitching, and an inability to move the arms, legs, and body. The condition continues to get worse. When the muscles in the chest area stop working, it becomes hard or impossible to breathe.
ALS affects approximately 7 out of every 100,000 people worldwide.
Having a family member who has a hereditary form of the disease is a risk factor for ALS. Other risks include military service. The reasons for this are unclear, but it may have to do with environmental exposure to toxins.
Symptoms
Symptoms usually do not develop until after age 50, but they can start in younger people. People with ALS have a loss of muscle strength and coordination that eventually gets worse and makes it impossible for them to do routine tasks such as going up steps, getting out of a chair, or swallowing.
Weakness can first affect the arms or legs, or the ability to breathe or swallow. As the disease gets worse, more muscle groups develop problems.
ALS does not affect the senses (sight, smell, taste, hearing, touch). Most people are able to think normally, although a small number develop dementia, causing problems with memory.
Muscle weakness starts in one body part, such as the arm or hand, and slowly gets worse until it leads to the following:
- Difficulty lifting, climbing stairs, and walking
- Difficulty breathing
- Difficulty swallowing -- choking easily, drooling, or gagging
- Head drop due to weakness of the neck muscles
- Speech problems, such as a slow or abnormal speech pattern (slurring of words)
- Voice changes, hoarseness
Other findings include:
- Depression
- Changing expressions of emotions (emotional lability)
- Muscle cramps
- Muscle stiffness, called spasticity
- Muscle contractions, called fasciculations
- Weight loss
Exams and Tests
Your health care provider will examine you and ask about your medical history and symptoms.
The physical exam may show:
- Weakness, often beginning in one area of the body
- Muscle tremors, spasms, twitching, or loss of muscle tissue
- Twitching of the tongue (common)
- Abnormal reflexes
- Stiff or clumsy walk
- Decreased or increased reflexes at the joints
- Difficulty controlling crying or laughing (sometimes called emotional incontinence)
- Loss of gag reflex
Tests that may be done include:
- Blood tests to check for other conditions
- Breathing test to see if lung muscles are affected
- Cervical spine CT or MRI to be sure there is no disease or injury to the neck, which can mimic ALS
- Electromyography (EMG) to see which nerves or muscles do not work properly
- Genetic testing, especially if there is a family history of ALS
- Head CT or MRI to rule out other conditions
- Swallowing studies
- Spinal tap (lumbar puncture)
Treatment
There is no known cure for ALS. Two medicines are available that help slow the progression of symptoms and may help people live slightly longer:
- Riluzole (Rilutek)
- Edaravone (Radicava)
Treatments to control other symptoms include:
- Baclofen or diazepam for spasticity that interferes with daily activities
- Trihexyphenidyl or amitriptyline for people with problems swallowing their own saliva
Trials of gene therapy are now underway for some specific hereditary causes.
Physical therapy, rehabilitation, use of braces or a wheelchair, or other measures may be needed to help with muscle function and general health. Ongoing oversight by a physical medicine provider is helpful, given continuously changing symptoms.
People with ALS tend to lose weight. The illness itself increases the need for food and calories. At the same time, problems with choking and swallowing make it hard to eat enough. To help with feeding, a tube may be placed into the stomach. A dietitian who specializes in ALS can give advice on healthy eating.
Breathing devices include machines that are considered non-invasive (such as CPAP or BiPAP). Others require tube in the trachea (invasive ventilation).
Medicine for depression may be needed if a person with ALS is diagnosed with depression. They also should discuss their wishes regarding artificial ventilation with their families and providers.
Support Groups
Emotional support is vital in coping with the disorder, because mental functioning is not affected. Groups such as the ALS Association may be available to help people who are coping with the disorder.
Support for people who are caring for someone with ALS is also available, and may be very helpful.
Outlook (Prognosis)
Over time, people with ALS lose the ability to function and care for themselves. Death often occurs within 3 to 5 years of diagnosis. About 1 in 4 people survive for more than 5 years after diagnosis. Some people live much longer, but they typically need help breathing from a ventilator or other device.
Possible Complications
Complications of ALS include:
- Breathing in food or fluid (aspiration)
- Loss of ability to care for self
- Lung failure
- Pneumonia
- Pressure sores
- Weight loss
When to Contact a Medical Professional
Contact your provider if:
- You have symptoms of ALS, particularly if you have a family history of the disorder
- You or someone else has been diagnosed with ALS and symptoms get worse or new symptoms develop
Increased difficulty swallowing, difficulty breathing, and episodes of apnea are symptoms that require immediate attention.
Prevention
You may want to see a genetic counselor if you have a family history of ALS.
Related Information
Community-acquired pneumonia in adultsRespiratory
Acute respiratory distress syndrome
References
Fearon C, Murray B, Mitsumoto H. Disorders of upper and lower motor neurons. In: Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Philadelphia, PA: Elsevier; 2022:chap 97.
Shaw PJ, Cudkowicz ME. Amyotrophic lateral sclerosis and other motor neuron diseases. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 387.
BACK TO TOPReview Date: 6/13/2024
Reviewed By: Joseph V. Campellone, MD, Department of Neurology, Cooper Medical School at Rowan University, Camden, NJ. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.

Health Content Provider
06/01/2025
|
A.D.A.M., Inc. is accredited by URAC, for Health Content Provider (www.urac.org). URAC's accreditation program is an independent audit to verify that A.D.A.M. follows rigorous standards of quality and accountability. A.D.A.M. is among the first to achieve this important distinction for online health information and services. Learn more about A.D.A.M.'s editorial policy, editorial process and privacy policy. A.D.A.M. is also a founding member of Hi-Ethics. This site complied with the HONcode standard for trustworthy health information from 1995 to 2022, after which HON (Health On the Net, a not-for-profit organization that promoted transparent and reliable health information online) was discontinued. |
The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. © 1997- 2025 A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited.
 All rights reserved.
All rights reserved.