
Síndrome de Noonan
Es una enfermedad presente al momento del nacimiento (congénita) que causa desarrollo anormal en muchas partes del cuerpo anormal. En aproximadamente el 50% de los casos se puede transmitir de padres a hijos (hereditaria).
Causas
El síndrome de Noonan está ligado a variantes en varios genes. En general, ciertas proteínas involucradas en el crecimiento y desarrollo se vuelvan hiperactivas como resultado de estos cambios genéticos.
El síndrome de Noonan es una afección autosómica dominante. Esto significa que solo uno de los padres tiene que aportar el gen variante para que el bebé tenga el síndrome. Sin embargo, es posible que algunos casos no sean hereditarios y en cambio se producen por casualidad (espontáneamente).
Autosómica dominante
Es una de varias formas en que un rasgo genético o trastorno se puede transmitir de padres a hijos. En una enfermedad autosómica dominante, si usted ...

Síntomas
Los síntomas incluyen:
- Retardo en la pubertad
- Ojos inclinados hacia abajo o muy separados
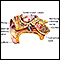
- Pérdida de la audición (varía)
Pérdida de la audición
Es la incapacidad total o parcial para escuchar sonidos en uno o ambos oídos.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Orejas de implantación baja o de forma anormal
- Discapacidad intelectual leve (solo en aproximadamente el 25% de los casos)
- Párpados caídos (ptosis)
Ptosis
La caída de párpado es el exceso de piel del párpado superior. El extremo del párpado superior puede llegar más abajo de lo que debería (ptosis) o p...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Estatura baja
Estatura baja
Un niño con estatura baja es mucho más bajo que otros niños de su misma edad y sexo. Su proveedor de atención médica le explicará la tabla de crecim...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Pene pequeño
- Testículos que no descienden
- Forma inusual del tórax (con frecuencia un tórax hundido llamado tórax excavado)
- Cuello con pliegues y de apariencia corta
Pruebas y exámenes
Su proveedor de atención médica llevará a cabo un examen físico. Este puede mostrar signos de problemas cardíacos que el bebé tenía desde el nacimiento. Pueden abarcar estenosis pulmonar y comunicación interauricular.
Estenosis pulmonar
Es un trastorno de válvula cardíaca que compromete la válvula pulmonar. Esta válvula separa el ventrículo derecho (una de las cámaras del corazón) de...
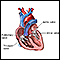
Comunicación interauricular
Es un defecto cardíaco que está presente al nacer (congénito). Mientras el bebé se desarrolla en el útero, se forma una pared (llamada tabique intera...

Los exámenes dependen de los síntomas, pero pueden incluir:
- Conteo de plaquetas
Conteo de plaquetas
Es un examen de laboratorio que mide la cantidad de plaquetas que usted tiene en la sangre. Las plaquetas son partículas en la sangre que ayudan a l...
Lea el artículo ahora Marcar el artículo como favorito - Examen de factores de coagulación de la sangre
- ECG, radiografía del tórax o ecocardiografía (ECHO)
ECG
Un electrocardiograma (ECG) es un examen que registra la actividad eléctrica del corazón.
 ImagenLea el artículo ahora Marcar el artículo como favorito
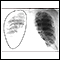
ImagenLea el artículo ahora Marcar el artículo como favoritoRadiografía del tórax
Es una radiografía del tórax, los pulmones, el corazón, las grandes arterias, las costillas y el diafragma.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favoritoEcocardiografía
Es un examen que utiliza ondas sonoras para crear imágenes del corazón. Dicha imagen, y la información que produce, son mucho más detalladas que una...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Audiometrías
- Niveles de hormona del crecimiento
- Ultrasonido renal
Las pruebas genéticas pueden ayudar a diagnosticar este síndrome.
Tratamiento
No hay un tratamiento específico. Su proveedor sugerirá un tratamiento para aliviar o manejar los síntomas. La hormona del crecimiento se ha utilizado con éxito en algunas personas con este síndrome para tratar la estatura baja.
Grupos de apoyo
Se puede encontrar más información y apoyo para las personas con el síndrome de Noonan y sus familias en -- www.teamnoonan.org y www.rasopathiesnet.org
Posibles complicaciones
Las complicaciones pueden incluir:
- Sangrado o anormal o formación de hematomas
- Acumulación de líquido en los tejidos corporales (linfedema, higroma quístico)
Higroma quístico
Es un tumor que a menudo se presenta en la zona de la cabeza y el cuello. Es una anomalía congénita.
Lea el artículo ahora Marcar el artículo como favorito - Retraso en el desarrollo en los bebés
- Leucemia y otros cánceres
Leucemia
La leucemia es un tipo de cáncer de la sangre que comienza en la médula ósea, el tejido blando que se encuentra en el centro de los huesos, donde se ...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Autoestima baja
- Infertilidad masculina si ambos testículos no han descendido
- Problemas con la estructura y función del corazón
- Estatura baja
- Problemas sociales debido a los síntomas físicos
Cuándo contactar a un profesional médico
Esta afección se puede detectar en los primeros exámenes que se le hacen al bebé. Con frecuencia, se necesita un genetista para diagnosticar este síndrome.
Prevención
Es posible que las parejas con antecedentes personales o familiares de síndrome de Noonan quieran solicitar asesoría genética antes de tener hijos.
Asesoría genética
La genética es el estudio de la herencia, el proceso en el que un padre les transmite ciertos genes a sus hijosLa apariencia de una persona, como su ...

Referencias
Cooke DW, Divall SA, Radovick S. Normal and aberrant growth in children. In: Melmed S, Auchus RJ, Goldfine AB, Koenig RJ, Rosen CJ, eds. Williams Textbook of Endocrinology. 14th ed. Philadelphia, PA: Elsevier; 2020:chap 25.
Madan-Khetarpal S, Arnold G, Ortiz D. Genetic disorders and dysmorphic conditions. In: Zitelli BJ, McIntire SC, Nowalk AJ, Garrison J, eds. Zitelli and Davis' Atlas of Pediatric Physical Diagnosis. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 1.
Mitchell AL. Congenital anomalies. In: Martin RJ, Fanaroff AA, Walsh MC, eds. Fanaroff and Martin's Neonatal-Perinatal Medicine. 12th ed. Philadelphia, PA: Elsevier; 2025:chap 29.
Pectus excavatum - ilustración
El pectus excavatum es una condición en la cual el esternón luce hendido y, el tórax, cóncavo. Algunas veces se le llama pecho en embudo. La mayoría de los casos corresponde a hallazgos aislados, es decir, que no se asocian con ninguna otra condición. Sin embargo, el pectus excavatum forma parte de algunas condiciones genéticas.
Pectus excavatum
ilustración

Pectus excavatum - ilustración
El pectus excavatum es una condición en la cual el esternón luce hendido y, el tórax, cóncavo. Algunas veces se le llama pecho en embudo. La mayoría de los casos corresponde a hallazgos aislados, es decir, que no se asocian con ninguna otra condición. Sin embargo, el pectus excavatum forma parte de algunas condiciones genéticas.
Pectus excavatum
ilustración
Actualizado: 3/31/2024
Versión en inglés revisada por: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 Todos los derechos son reservados
Todos los derechos son reservados