Mucopolisacaridosis tipo I
La mucopolisacaridosis tipo I (MPS I) es una rara enfermedad en la cual el cuerpo carece o no tiene suficiente cantidad de una enzima necesaria para descomponer cadenas largas de moléculas de azúcar. Estas cadenas son llamadas glucosaminoglucanos (anteriormente denominados mucopolisacáridos). Como resultado, las moléculas se acumulan en diferentes partes del cuerpo y causan diversos problemas de salud.
Enzima
Las enzimas son proteínas complejas que producen un cambio químico específico. Por ejemplo, pueden ayudar a descomponer los alimentos que consumimos...
Lea el artículo ahora Marcar el artículo como favoritoEsta afección pertenece a un grupo de enfermedades llamado mucopolisacaridosis (MPS). MPS I es el tipo más común
Mucopolisacaridosis
MPS son un grupo de enfermedades inusuales en las cuales el cuerpo presenta ausencia o no tiene suficiente de una enzima necesaria para descomponer l...
Lea el artículo ahora Marcar el artículo como favoritoExisten varios tipos de MPS, incluyendo:
- MPS II (síndrome de Hunter)
MPS II
La mucopolisacaridosis tipo II (MPS II) es una enfermedad rara en la cual al cuerpo le falta o no tiene suficiente cantidad de una enzima necesaria p...
Lea el artículo ahora Marcar el artículo como favorito - MPS III (síndrome de Sanfilippo)
MPS III
MPS III es una enfermedad inusual en la cual el cuerpo presenta ausencia o no tiene suficiente de ciertas enzimas necesarias para descomponer las cad...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - MPS IV (síndrome de Morquio)
MPS IV
La mucopolisacaridosis tipo IV (MPS IV) es una enfermedad rara en la cual el cuerpo carece o no tiene suficiente cantidad de una enzima necesaria par...
Lea el artículo ahora Marcar el artículo como favoritoMPS son un grupo de enfermedades inusuales en las cuales el cuerpo presenta ausencia o no tiene suficiente de una enzima necesaria para descomponer l...
Lea el artículo ahora Marcar el artículo como favorito
Causas
La MPS I es hereditaria, lo cual significa que sus padres normalmente le transmiten la enfermedad a usted. Si ambos padres portan una copia inactiva del gen relacionado con esta afección, cada uno de sus hijos tiene un 25% (1 en 4) de probabilidades de desarrollar la enfermedad.
Las personas con MPS I no producen una enzima llamada alfa-L-iduronidasa lisosómica. Esta enzima ayuda a descomponer las cadenas largas de moléculas de azúcar llamadas glucosaminoglucanos. Estas moléculas se encuentran en todo el cuerpo, a menudo en las secreciones mucosas y en el líquido que rodea las articulaciones.
Sin la enzima, los glucosaminoglucanos se acumulan y causan daño a órganos, incluso al corazón. Los síntomas pueden ir de leves a graves. La forma leve se denomina MPS I atenuada y la forma severa se denomina MPS I grave.
Síntomas
Los síntomas de MPS I casi siempre aparecen de los 3 a los 8 años de edad. Los niños con MPS I grave presentan síntomas antes que aquellos con la forma menos grave.
Algunos de los síntomas incluyen:
- Huesos anormales en la columna
- Incapacidad de abrir completamente la mano (mano en garra)
Mano en garra
Es una afección que ocasiona que los dedos se curven o se doblen. Esto le da a la mano la apariencia de la garra de un animal.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Córneas opacas
Córneas opacas
Es la pérdida de transparencia de la córnea.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Sordera
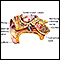
Sordera
Es la incapacidad total o parcial para escuchar sonidos en uno o ambos oídos.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Crecimiento interrumpido
- Problemas de válvula cardíaca
- Enfermedad articular, incluso rigidez
- Discapacidad intelectual que empeora con el tiempo en la MPS I grave
Discapacidad intelectual
Es una afección diagnosticada antes de los 18 años de edad que incluye un funcionamiento intelectual general por debajo del promedio y una carencia d...
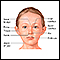
Lea el artículo ahora Marcar el artículo como favorito - Rasgos faciales gruesos y toscos con puente nasal bajo
Puente nasal bajo
Es el aplanamiento de la parte superior de la nariz.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito
Pruebas y exámenes
En algunos estados, a los bebés se les examina en busca de MPS I como parte de las pruebas de detección para recién nacidos.
Pruebas de detección para recién nacido
Las pruebas de detección (tamizaje) para recién nacidos buscan trastornos metabólicos, genéticos y del desarrollo en bebés recién nacidos. Esto perm...
Lea el artículo ahora Marcar el artículo como favoritoOtros exámenes que se pueden hacer dependiendo de los síntomas incluyen:
-
Electrocardiograma ECG
ECG
Un electrocardiograma (ECG) es un examen que registra la actividad eléctrica del corazón.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Pruebas genéticas para buscar cambios en el gen de la alfa-L-iduronidasa (IDUA)
- Exámenes de orina en busca de mucopolisacáridos adicionales
- Radiografía de la columna
Tratamiento
La terapia de reemplazo enzimático se puede recomendar. El medicamento llamado laronidasa (Aldurazyme), se administra a través de una vena (IV, vía intravenosa). Reemplaza la carencia de la enzima. Hable con el proveedor de su hijo para obtener más información.
Se ha utilizado un trasplante de médula ósea. El tratamiento ha tenido resultados mixtos.
Trasplante de médula ósea
Es un procedimiento para reemplazar la médula ósea dañada o enferma con células madre de médula ósea sana. La médula ósea es el tejido graso y blando...

Otros tratamientos dependen de los órganos que están afectados.
Grupos de apoyo
Se puede encontrar más información y apoyo para las personas con MPS I y sus familias en:
- National MPS Society -- www.mpssociety.org
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/mucopolysaccharidosis-type-i
- NIH Genetic and Rare Disorders Information Center -- rarediseases.info.nih.gov/diseases/10335/mucopolysaccharidosis-type-i
Expectativas (pronóstico)
Los niños con MPS I grave por lo general no tienen un buen pronóstico. Sus problemas de salud empeoran con el tiempo, resultando en la muerte a la edad de 10 años.
Los niños con MPS I atenuada (más leve) tienen menos problemas de salud, muchos con vidas bastante normales que llegan a la adultez.
Cuándo contactar a un profesional médico
Comuníquese con su proveedor si:
- Tiene antecedentes familiares de MPS I y está considerando la posibilidad de tener hijos
- Su hijo empieza a mostrar síntomas de MPS I
Prevención
Los expertos recomiendan la asesoría genética y la realización de exámenes para parejas con antecedentes familiares de MPS I que estén contemplando la posibilidad de tener hijos. Hay disponibilidad de pruebas prenatales.
Asesoría genética
La genética es el estudio de la herencia, el proceso en el que un padre les transmite ciertos genes a sus hijosLa apariencia de una persona, como su ...

Revisado por
Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 239.
Spranger JW. Mucopolysaccharidoses. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 107.
Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18.

 Todos los derechos son reservados.
Todos los derechos son reservados.