
Atrofia multisistémica - tipo parkinsoniano
Síndrome de Shy-Drager; Hipotensión ortostática neurológica; Síndrome de Shy-McGee-Drager; Síndrome de Parkinson plus; AMS-P; AMS-CLa atrofia multisistémica - tipo parkinsoniano (AMS-P) es una afección poco frecuente que causa síntomas similares al mal de Parkinson. Sin embargo, las personas con esta enfermedad presentan un daño más generalizado a la parte del sistema nervioso que controla funciones importantes, como la frecuencia cardíaca, la presión arterial y la sudoración.
Mal de Parkinson
El mal de Parkinson provoca la muerte de ciertas células del cerebro, que son las que ayudan a controlar el movimiento y la coordinación. La enferme...

El otro subtipo de atrofia multisistémica (AMS) es la AMS - cerebelar. Afecta principalmente el cerebelo, un área profunda del cerebro, justo sobre la médula espinal.
AMS - cerebelar
La atrofia multisistémica del tipo cerebelosa (AMS-C) es una enfermedad poco frecuente que hace que ciertas zonas profundas en el cerebro, justo por ...
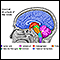
Causas
La causa de la AMS-P se desconoce. Las áreas afectadas del cerebro se traslapan con las áreas afectadas por la enfermedad de Parkinson, con síntomas similares. Por esta razón, este subtipo de AMS es llamado tipo parkinsoniano.
La AMS-P con mucha frecuencia se diagnostica en hombres mayores de 60 años.
Síntomas
La AMS daña al sistema nervioso. La enfermedad tiende a progresar rápidamente. Aproximadamente la mitad de las personas con AMS-P pierde la mayoría de sus habilidades motrices en un período de 5 años después de que empezó la enfermedad.
Los síntomas pueden incluir:
- Temblores
- Problemas de movimiento como lentitud, pérdida del balance, arrastrar los pies al caminar
- Caídas frecuentes
- Molestia y dolor muscular (mialgia) y rigidez
- Cambios faciales, como cara con apariencia de máscara y mirada fija
- Dificultad para masticar o tragar (ocasionalmente), incapaz de cerrar la boca
- Interrupción en los patrones del sueño (frecuentemente durante el sueño con movimientos oculares rápidos [MOR] tarde en la noche)
- Mareos o desmayos al estar de pie o después de pararse
-
Problemas para tener erecciones
Problemas para tener erecciones
Un problema de erección se produce cuando un hombre no puede lograr o mantener una erección que sea lo suficientemente firme para una relación sexual...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Pérdida de control intestinal o vesical (heces u orina)
- Dificultad con cualquier actividad que requiera movimientos pequeños (pérdida de las destrezas motoras finas), como una escritura pequeña e ilegible
- Pérdida de sudoración de cualquier parte del cuerpo
- Disminución de la función intelectual
- Náuseas y problemas con la digestión
- Dificultades en la postura, como puede ser inestable, encorvada, inclinada hacia adelante
-
Cambios en la visión, disminución o visión borrosa
Cambios en la visión
Existen muchos tipos de problemas oculares y perturbaciones visuales como: Halos Visión borrosa (pérdida de la agudeza visual y la incapacidad de ver...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Cambios en la voz y el habla
Otros síntomas que se pueden presentar con esta enfermedad:
-
Confusión
Confusión
Es la incapacidad para pensar de manera tan clara y rápida como uno normalmente lo hace. Usted puede sentirse desorientado y tener dificultad para p...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito -
Demencia
Demencia
Es una pérdida de la función cerebral que ocurre a causa de ciertas enfermedades. Esto afecta a una o más funciones cerebrales como la memoria, el p...
ImagenLea el artículo ahora Marcar el artículo como favorito -
Depresión
Depresión
La depresión se puede describir como el hecho de sentirse triste, melancólico, infeliz, abatido o derrumbado. La mayoría de nosotros se siente de es...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Dificultad para respirar relacionada con el sueño, incluyendo apnea del sueño y obstrucción de las vías respiratorias, que lleva a un sonido vibrante bronco
Apnea del sueño
La apnea obstructiva del sueño (AOS) es un problema que ocurre cuando la respiración se detiene mientras usted está dormido. Esto ocurre porque las ...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Piernas inquietas
Pruebas y exámenes
Su proveedor de atención médica lo examinará y le revisará los ojos, los nervios y los músculos.
Le medirán la presión arterial mientras está acostado y parado.
No existen exámenes específicos para confirmar esta enfermedad. Un médico especializado en el sistema nervioso (neurólogo) puede hacer el diagnóstico basándose en:
- El historial de los síntomas
- Los resultados del examen físico
- Exclusión de otras causas de los síntomas
Los exámenes para ayudar a confirmar el diagnóstico pueden incluir:
-
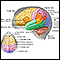
Resonancia magnética de la cabeza
Resonancia magnética de la cabeza
Una RM (resonancia magnética) de la cabeza es un examen imagenológico que utiliza imanes y ondas de radio potentes para crear imágenes del cerebro y ...
ImagenLea el artículo ahora Marcar el artículo como favorito - Niveles de norepinefrina en el plasma
Norepinefrina
Es una prueba que mide los niveles de catecolaminas en la sangre. Las catecolaminas son hormonas producidas por las glándulas suprarrenales. Las ca...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Examen de orina para los productos de la descomposición de la norepinefrina (catecolaminas en orina)
Catecolaminas en orina
Las catecolaminas son químicos producidos por el tejido nervioso (incluso el cerebro) y la glándula suprarrenal. Los tipos principales de catecolamin...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito
Tratamiento
No existe una cura para la AMS-P. Tampoco una forma conocida para evitar que empeore. El objetivo del tratamiento es controlar los síntomas.
Se pueden utilizar medicamentos dopaminérgicos, como levodopa y carbidopa, para reducir los temblores iniciales o leves.
Sin embargo, para la mayoría de las personas con AMS-P, estos medicamentos no funcionan bien.
Se pueden utilizar medicamentos para tratar la presión arterial baja.
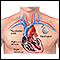
Un marcapasos programado para estimular al corazón a latir a un ritmo rápido (más de 100 latidos por minuto) puede aumentar la presión arterial para algunas personas.
Marcapasos
Es un pequeño dispositivo operado con pilas. Percibe cuándo el corazón está latiendo en forma muy lenta. Este envía una señal al corazón, la cual l...
El estreñimiento se puede tratar con una dieta rica en fibra y laxantes. Hay medicamentos disponibles para tratar la impotencia.
Grupos de apoyo
Puede encontrar más información y apoyo para personas con AMS-P y sus familias en:
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/multiple-system-atrophy/
- The MSA Coalition -- missionmsa.org
Expectativas (pronóstico)
El desenlace clínico del AMS es desalentador. La pérdida de las funciones mentales y físicas empeora lentamente. Es probable que se produzca una muerte prematura. Los pacientes normalmente viven de 7 a 9 años después del diagnóstico.
Cuándo contactar a un profesional médico
Comuníquese con su proveedor si presenta síntomas de este trastorno.
Comuníquese con su proveedor si ya le han diagnosticado esta enfermedad y los síntomas reaparecen o empeoran. También comuníquese con su proveedor si aparecen nuevos síntomas, incluso posibles efectos secundarios de los medicamentos, como:
- Cambios en la lucidez mental/conducta/estado de ánimo
- Conducta delirante
- Vértigo
-
Alucinaciones
Alucinaciones
Consisten en percibir cosas como visiones, sonidos u olores que parecen reales, pero no lo son. Estas cosas son creadas por la mente.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Movimientos involuntarios
- Pérdida de funciones mentales
- Náuseas o vómitos
- Confusión o desorientación grave
Si tiene un familiar con AMS y su estado empeora hasta el punto en que ya no se lo puede seguir cuidando en el hogar, busque el consejo del proveedor de su familiar.
Referencias
Jankovic J. Parkinson disease and other movement disorders. In: Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Philadelphia, PA: Elsevier; 2022:chap 96.
National Institutes of Health website. National Institute of Neurological Disorders and Stroke. Multiple system atrophy. www.ninds.nih.gov/health-information/disorders/multiple-system-atrophy. Updated July 19, 2024. Accessed November 4, 2024.
Romero-Ortuno R, Wilson KJ, Hampton JL. Disorders of the autonomic nervous system. In: Fillit HM, Rockwood K, Young J, eds. Brocklehurst's Textbook of Geriatric Medicine and Gerontology. 8th ed. Philadelphia, PA: Elsevier; 2017:chap 63.
-
Sistema nervioso central y sistema nervioso periférico - ilustración
El sistema nervioso central comprende el cerebro y la médula espinal. El sistema nervioso periférico incluye los nervios fuera del cerebro y la médula espinal.
Sistema nervioso central y sistema nervioso periférico
ilustración

Actualizado: 10/23/2024
Versión en inglés revisada por: Joseph V. Campellone, MD, Department of Neurology, Cooper Medical School at Rowan University, Camden, NJ. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 Todos los derechos son reservados
Todos los derechos son reservados