
Síndrome de Alport
Nefritis hereditaria; Hematuria - nefropatía - sordera; Nefritis hemorrágica familiar; Sordera hereditaria y nefropatíaEl síndrome de Alport es un trastorno hereditario poco frecuente que causa daño a los diminutos vasos sanguíneos en los riñones. También puede causar pérdida de la audición y problemas oculares.
Causas
El síndrome de Alport es una forma hereditaria de inflamación del riñón (nefritis). Es causado por un defecto (mutación) en un gen para una proteína en el tejido conectivo, llamada colágeno.
El trastorno poco común. Existen tres tipos genéticos:
- Síndrome de Alport ligado al cromosoma X (XLAS, por sus siglas en inglés): Este es el tipo más común. La enfermedad es más grave en hombres que en mujeres.
- Síndrome de Alport autosómico recesivo (ARAS, por sus siglas en inglés): La enfermedad es igual de grave en hombres y mujeres.
- Síndrome de Alport autosómico dominante (ADAS, por sus siglas en inglés): Este es el tipo menos frecuente. La enfermedad es igual de grave en hombres y mujeres.
En todos los tipos de síndrome de Alport, los riñones resultan afectados. Los diminutos vasos sanguíneos en los glomérulos de los riñones resultan dañados. Los glomérulos filtran la sangre para producir orina y eliminar los productos de desecho de la sangre.
Al principio, no hay síntomas. Con el tiempo, a medida que los glomérulos resultan más y más dañados, se pierde la función renal y hay una acumulación de líquidos y productos de desecho en el cuerpo. La afección puede progresar hasta convertirse en enfermedad renal terminal (ERT) a una edad temprana, entre la adolescencia y los 40 años de edad. En ese punto, es necesario someterse a diálisis o a un trasplante de riñón.
ERT
La enfermedad renal terminal (ERT), es la última etapa de la enfermedad renal crónica. Esto es cuando sus riñones ya no pueden atender las necesidad...
Diálisis
Con la diálisis se trata la enfermedad renal terminal también llamada disfunción renal. Este procedimiento elimina los residuos de la sangre cuando ...
Lea el artículo ahora Marcar el artículo como favoritoTrasplante de riñón
Es una cirugía para colocar un riñón sano en una persona con insuficiencia renal.
Síntomas
El síntoma principal del síndrome de Alport es sangre en la orina que con frecuencia no es visible y solo se detecta bajo un microscopio. Esto sucede a temprana edad. A medida que la enfermedad avanza, pueden aparecer otros síntomas.
RIÑONES
Otros síntomas de problemas de riñón incluyen:
- Color de orina anormal
Color de orina anormal
El color normal de la orina es amarillo paja. La orina de color anormal puede ser turbia, oscura o de color sangre.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Sangre en la orina que es visible al tener un resfriado o gripe o al hacer ejercicio
- Dolor de costado
- Presión arterial alta
- Hinchazón en todo el cuerpo (edema)
Hinchazón en todo el cuerpo
Es el agrandamiento de órganos, piel u otras partes del cuerpo, causado por la acumulación de líquidos en los tejidos. La presencia de líquido extra...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Fatiga
- Falta de apetito
- Sed excesiva
OÍDOS
Con el tiempo, el síndrome de Alport también causa pérdida de la audición de ambos oídos. Es común que en hombres con XLAS se haya presentado al inicio de la adolescencia, mientras que, en las mujeres, la pérdida de la audición no es tan común y sucede en la adultez. En el caso de ARAS, los niños, hombres y mujeres presentan pérdida de la audición durante la infancia. Con ADAS, se presenta más adelante en la vida.
La pérdida de la audición por lo regular ocurre antes que la insuficiencia renal.
OJOS
El síndrome de Alport también causa problemas oculares en quienes tienen XLAS y ARAS, que incluyen:
- Una forma anormal del cristalino (lenticono anterior), la cual puede causar un deterioro lento de la vista, así como cataratas.
Cataratas
Una catarata es una opacidad del cristalino del ojo.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Erosión corneal, en la que hay un daño de la capa exterior de la cubierta del glóbulo ocular, lo que provoca dolor, picazón, enrojecimiento del ojo, o visión borrosa.
- Coloración anormal de la retina, una afección denominada retinopatía punto-mancha. Esto no causa problemas para la visión, pero puede ayudar a diagnosticar el síndrome de Alport.
Retina
Es la capa de tejido sensible a la luz que se encuentra en la parte posterior globo ocular. Las imágenes que pasan a través del cristalino del ojo s...
ImagenLea el artículo ahora Marcar el artículo como favorito - Agujero macular, en el que hay un adelgazamiento o un rompimiento de la mácula. La mácula es la parte de la retina que hace que la visión central sea nítida y más detallada. Un agujero macular provoca visión central borrosa o distorsionada.
Los problemas oculares son poco comunes en personas con ADAS.
Pruebas y exámenes
El proveedor de atención médica lo examinará y hará preguntas sobre sus síntomas.
Se pueden hacer los siguientes exámenes:
- BUN y creatinina sérica
BUN
BUN (por sus siglas en inglés) corresponde a nitrógeno ureico en la sangre. El nitrógeno ureico es lo que se forma cuando la proteína se descompone....
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favoritoCreatinina
Es un análisis que mide el nivel de creatinina en la sangre. Se hace para ver qué tan bien están funcionando los riñones. La creatinina en la orina ...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Conteo sanguíneo completo (CSC)
Conteo sanguíneo completo (CSC)
Un hemograma o conteo sanguíneo completo (CSC) mide lo siguiente:La cantidad de glóbulos blancos (conteo de GB)La cantidad de glóbulos rojos (conteo ...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Biopsia renal
Biopsia renal
Es la extracción de un pequeño fragmento de tejido del riñón para su análisis.
ImagenLea el artículo ahora Marcar el artículo como favorito - Análisis de orina
Análisis de orina
Es la evaluación física, química y microscópica de la orina. Dicho análisis consta de varios exámenes para detectar y medir diversos compuestos que ...
ImagenLea el artículo ahora Marcar el artículo como favorito
Si su proveedor sospecha que usted tiene síndrome de Alport, probablemente también le realizarán exámenes de la visión y la audición.
Tratamiento
Los objetivos del tratamiento incluyen monitorear y controlar la enfermedad y tratar los síntomas.
Su proveedor puede recomendar cualquiera de los siguientes:
- Una dieta que limite el consumo de sal, líquidos y potasio
- Medicamentos para controlar la presión arterial alta
La enfermedad renal se maneja al:
- Tomar medicamentos para frenar el daño a los riñones
- Una dieta que limite el consumo de sal, líquidos y proteína
La pérdida de la audición se puede manejar con audífonos. Los problemas oculares se tratan según resulte necesario. Por ejemplo, un cristalino anormal a causa de un lenticono o de cataratas se puede reemplazar.
Se puede recomendar la asesoría genética debido a que el trastorno es hereditario.
Grupos de apoyo
Puede encontrar más información y grupos de apoyo para personas con síndrome de Alport y sus familias en:
- Alport Syndrome Foundation: alportsyndrome.org/for-patients/patient-resources/
- National Kidney Foundation: www.kidney.org/atoz/content/alport
- National Organization for Rare Disorders: rarediseases.org/rare-diseases/alport-syndrome
Expectativas (pronóstico)
El pronóstico depende del tipo de síndrome de Alport, sexo biológico y edad.
Tanto para hombres como para mujeres, la función renal decaerá con el tiempo. En hombres con XLAS, esto puede causar disfunción renal a una edad temprana. La mayoría de los hombres con XLAS experimentarán disfunción renal a la edad de 60 años. Las mujeres con XLAS pueden o no presentar problemas de riñón, pero el riesgo aumenta con la edad.
Tanto hombres como mujeres con ARAS desarrollarán disfunción renal a principios de la edad adulta.
El ADAS avanza lentamente tanto en hombres como en mujeres, y la disfunción renal puede no presentarse hasta más adelante en la vida.
A medida que los riñones fallan, será necesario realizar diálisis o trasplante de riñón. El trasplante de riñón es con frecuencia muy exitoso en personas con el síndrome de Alport.
Cuándo contactar a un profesional médico
Comuníquese con su proveedor para solicitar una cita si:
- Tiene síntomas del síndrome de Alport.
- Tiene un antecedente familiar de este síndrome y está planeando quedar embarazada.
- Su producción de orina disminuye o cesa o si observa sangre en la orina (esto puede ser un síntoma de enfermedad renal crónica).
Prevención
Tener conciencia de los factores de riesgo, tales como un antecedente familiar del problema, puede permitir que la afección se detecte en forma temprana.
Referencias
Gregory MC. Alport syndrome and related disorders. In: Gilbert SJ, Weiner DE, Bomback AS, Perazella MA, Rifkin DE, eds. National Kidney Foundation's Primer on Kidney Diseases. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 41.
Radhakrishnan J, Appel GB, D'Agati VD. Secondary glomerular disease. In: Yu ASL, Chertow GM, Luyckx VA, Marsden PA, Skorecki K, Taal MW, eds. Brenner and Rector's The Kidney. 11th ed. Philadelphia, PA: Elsevier; 2020:chap 32.
Rheault MN, Kashtan CE. Alport syndrome and other familial glomerular syndromes. In: Johnson RJ, Floege J, Tonelli M, eds. Comprehensive Clinical Nephrology. 7th ed. Philadelphia, PA: Elsevier; 2024:chap 48.
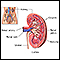
Anatomía del riñón - ilustración
Los riñones son responsables de eliminar los desechos del cuerpo, regular el equilibrio electrolítico y estimular la producción de glóbulos rojos.
Anatomía del riñón
ilustración

Actualizado: 5/19/2023
Versión en inglés revisada por: Walead Latif, MD, Nephrologist and Clinical Associate Professor, Rutgers Medical School, Newark, NJ. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 Todos los derechos son reservados
Todos los derechos son reservados