
Síndrome de Marfan
Aneurisma aórtica - MarfanEs un trastorno del tejido conectivo. Este es el tejido fortalece las estructuras corporales.
Los trastornos del tejido conectivo afectan los sistemas esquelético y cardiovascular, al igual que los ojos y la piel.
Causas
El síndrome de Marfan es causado por variantes en un gen llamado fibrilina-1. Este gen juega un papel importante como pilar fundamental para el tejido conectivo en el cuerpo.
El variante en el gen también causa crecimiento excesivo de los huesos largos del cuerpo. Las personas con este síndrome tienen estatura elevada y las piernas y manos largas. La forma como ocurre este crecimiento exagerado no se ha comprendido bien.
Otras áreas del cuerpo que resultan afectadas incluyen:
- El tejido pulmonar (puede haber un neumotórax, en el cual el aire se puede escapar del pulmón hacia la cavidad torácica y colapsar el pulmón)
- La aorta, el principal vaso sanguíneo que lleva sangre del corazón al cuerpo, puede estirarse o debilitarse (lo que se denomina dilatación aórtica o aneurisma aórtico)
- Las válvulas cardíacas
- Los ojos, provocando cataratas y otros problemas (como por ejemplo, luxación del cristalino)
- La piel
- El tejido que cubre la médula espinal
- Las articulaciones
En la mayoría de los casos, el síndrome de Marfan se transmite de padres a hijos (hereditario). Sin embargo, hasta el 30% de las personas no tiene un antecedente familiar, lo cual se denomina "esporádico". En los casos esporádicos, se cree que el síndrome es ocasionado por un nuevo cambio genético.
Síntomas
Las personas con el síndrome de Marfan generalmente son altas con brazos y piernas delgados y largos, y dedos en forma de araña (llamado aracnodactilia). Cuando estiran los brazos, la longitud de éstos es mayor que su estatura.
Otros síntomas incluyen:
- Un tórax que se hunde o sobresale, llamado tórax en embudo (tórax excavado) o pecho de paloma (tórax en quilla)
Tórax excavado
Es un término médico que describe una formación anómala de la caja torácica que le da al pecho una apariencia hundida o deprimida.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favoritoTórax en quilla
Se presenta cuando el pecho protruye sobre el esternón. A menudo se dice que le da a la persona una apariencia de pájaro.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Pie plano
- Paladar muy arqueado y dientes apiñados
-
Hipotonía de los músculos (bajo tono muscular)
Hipotonía
Significa disminución del tono muscular.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Articulaciones que son demasiado flexibles (pero los codos pueden ser menos flexibles)
- Dificultades de aprendizaje
- Movimiento del cristalino del ojo de su posición normal (luxación)
- Miopía
- Mandíbula pequeña y baja (micrognatia)
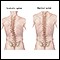
- Columna que se curva hacia un lado (escoliosis)
Escoliosis
Es una curvatura anormal de la espina dorsal. Esta es su columna vertebral. El hueso que baja por la espalda. La columna vertebral de toda persona...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Cara estrecha y delgada
Muchas personas con síndrome de Marfan sufren de dolores crónicos en los músculos y las articulaciones.
Pruebas y exámenes
El Proveedor de atención médica llevará a cabo un examen físico. Las articulaciones se pueden mover más de lo normal. También puede haber signos de:
-
Aneurisma
Aneurisma
Un aneurisma es una dilatación o ensanchamiento anormal de una porción de una arteria, debido a una debilidad de la pared del vaso sanguíneo. Un aneu...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Atelectasia pulmonar
- Problemas con las válvulas cardíacas
Un examen ocular puede mostrar:
- Defectos del cristalino o la córnea
-
Desprendimiento de retina
Desprendimiento de retina
Es la separación de la membrana sensible a la luz (retina) en la parte posterior del ojo de sus capas de soporte.
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Problemas visuales
Se pueden llevar a cabo los siguientes exámenes:
-
Ecocardiografía
Ecocardiografía
Es un examen que utiliza ondas sonoras para crear imágenes del corazón. Dicha imagen, y la información que produce, son mucho más detalladas que una...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Pruebas de la mutación de la fibrilina-1 (en algunas personas)
Cada año se debe realizar una ecocardiografía u otro examen para observar la base de la aorta y posiblemente las válvulas cardíacas. Dependiendo de los resultados, es posible que necesite esta prueba con menos frecuencia que una vez al año.
Tratamiento
Los problemas visuales se deben tratar cuando sea posible.
Vigile la escoliosis, especialmente durante los años de la adolescencia.
Los medicamentos para disminuir la frecuencia cardíaca y bajar la presión arterial pueden ayudar a prevenir el estrés en la aorta. Para evitar lesionar la aorta, las personas con esta afección pueden tener que cambiar sus actividades. Algunas personas pueden necesitar cirugía para reemplazar la válvula y la raíz aórtica.
Se debe vigilar muy de cerca a las mujeres embarazadas que tengan este síndrome, debido al incremento de la tensión sobre el corazón y la aorta.
Grupos de apoyo
Puede encontrar más información y apoyo para personas con síndrome de Marfan y sus familias en:
La Fundación Marfan -- marfan.org
Expectativas (pronóstico)
Las complicaciones relacionadas con el corazón pueden acortar el período de vida de las personas con esta enfermedad. Sin embargo, muchas personas viven hasta los 60 años y más. El período de vida se puede extender más con un buen cuidado y cirugía.
Posibles complicaciones
Las complicaciones pueden incluir:
-
Regurgitación aórtica
Regurgitación aórtica
Es una valvulopatía cardíaca en la cual la válvula aórtica no se cierra herméticamente. Esto permite que la sangre fluya desde la aorta (el vaso san...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Ruptura aórtica
- Endocarditis bacteriana
-
Aneurisma aórtico disecante (también llamado disección aórtica)
Aneurisma aórtico disecante
Es una afección grave en la cual hay una ruptura en la pared de la arteria principal que transporta la sangre fuera del corazón (la aorta). A medida...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Agrandamiento de la base de la aorta
-
Insuficiencia cardíaca
Insuficiencia cardíaca
La insuficiencia cardíaca es una afección en la cual el corazón ya no puede bombear sangre rica en oxígeno al resto del cuerpo de forma eficiente. E...
 ImagenLea el artículo ahora Marcar el artículo como favorito
ImagenLea el artículo ahora Marcar el artículo como favorito - Prolapso de la válvula mitral
- Escoliosis
- Problemas de visión
Cuándo contactar a un profesional médico
Las parejas que tengan esta afección y estén planeando tener hijos posiblemente necesiten consultar con un asesor en genética antes de tener familia.
Prevención
Las variantes de genes nuevas y espontáneas que llevan al síndrome de Marfan (menos de un tercio de los casos) no se pueden prevenir. Si usted padece el síndrome de Marfan, vea a su proveedor al menos una vez cada año.
Referencias
Doyle JJ, Dietz HC. Marfan syndrome. In: Kliegman RM, St. Geme JW, Blum NJ, et al. eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 743.
Madan-Khetarpal S, Arnold G, Ortiz D. Genetic disorders and dysmorphic conditions. In: Zitelli BJ, McIntire SC, Nowalk AJ, Garrison J, eds. Zitelli and Davis' Atlas of Pediatric Diagnosis. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 1.
Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 239.
-
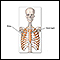
Pectus excavatum - ilustración
El pectus excavatum es una condición en la cual el esternón luce hendido y, el tórax, cóncavo. Algunas veces se le llama pecho en embudo. La mayoría de los casos corresponde a hallazgos aislados, es decir, que no se asocian con ninguna otra condición. Sin embargo, el pectus excavatum forma parte de algunas condiciones genéticas.
Pectus excavatum
ilustración
-
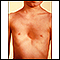
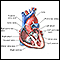
Síndrome de Marfan - ilustración
El síndrome de Marfan es un trastorno del tejido conectivo el cual causa defectos esqueléticos que se reflejan en el típico aspecto alto y larguirucho que presenta la persona. En ella pueden observarse extremidades largas y dedos en forma de araña, anomalías en el pecho, curvatura de la columna y un grupo particular de rasgos faciales que incluyen un paladar con arco muy elevado y dientes apiñados. El defecto más significativo en este síndrome es la presencia de anomalías cardiovasculares, entre las cuales está el agrandamiento o dilatación de la base de la aorta. Debido a que el síndrome de Marfan es usualmente un trastorno hereditario, los futuros padres que tengan antecedentes familiares de este síndrome deben buscar asesoramiento genético.
Síndrome de Marfan
ilustración


-
Pectus excavatum - ilustración
El pectus excavatum es una condición en la cual el esternón luce hendido y, el tórax, cóncavo. Algunas veces se le llama pecho en embudo. La mayoría de los casos corresponde a hallazgos aislados, es decir, que no se asocian con ninguna otra condición. Sin embargo, el pectus excavatum forma parte de algunas condiciones genéticas.
Pectus excavatum
ilustración
-
Síndrome de Marfan - ilustración
El síndrome de Marfan es un trastorno del tejido conectivo el cual causa defectos esqueléticos que se reflejan en el típico aspecto alto y larguirucho que presenta la persona. En ella pueden observarse extremidades largas y dedos en forma de araña, anomalías en el pecho, curvatura de la columna y un grupo particular de rasgos faciales que incluyen un paladar con arco muy elevado y dientes apiñados. El defecto más significativo en este síndrome es la presencia de anomalías cardiovasculares, entre las cuales está el agrandamiento o dilatación de la base de la aorta. Debido a que el síndrome de Marfan es usualmente un trastorno hereditario, los futuros padres que tengan antecedentes familiares de este síndrome deben buscar asesoramiento genético.
Síndrome de Marfan
ilustración
Actualizado: 5/8/2024
Versión en inglés revisada por: Thomas S. Metkus, MD, Assistant Professor of Medicine and Surgery, Johns Hopkins University School of Medicine, Baltimore, MD. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 Todos los derechos son reservados
Todos los derechos son reservados