
Gaucher disease
Glucocerebrosidase deficiency; Glucosylceramidase deficiency; Lysosomal storage disease - Gaucher; Gaucher's diseaseGaucher disease is a rare genetic disorder in which a person lacks an enzyme called glucocerebrosidase (GBA).
Causes
Gaucher disease is rare in the general population. People of Eastern and Central European (Ashkenazi) Jewish heritage are more likely to have this disease.
It is an autosomal recessive disease. This means that the mother and father must both pass one abnormal copy of the disease gene to their child in order for the child to develop the disease. A parent who carries an abnormal copy of the gene but doesn't have the disease is called a silent carrier.
Autosomal recessive
Autosomal recessive is one of several ways that a trait, disorder, or disease can be passed down through families. An autosomal recessive disorder me...

The lack of the GBA causes harmful substances to build up in the liver, spleen, bones, and bone marrow. These substances prevent cells and organs from working properly.
There are three main subtypes of Gaucher disease:
- Type 1 is most common. It involves bone disease, anemia, an enlarged spleen and low platelets (thrombocytopenia). Type 1 affects both children and adults. It is most common in the Ashkenazi Jewish population.
- Type 2 usually begins in infancy with severe neurologic involvement. This form can lead to rapid, early death.
- Type 3 may cause liver, spleen, and brain problems. People with this type may live into adulthood.
Symptoms
Bleeding because of low platelet count is the most common symptom seen in Gaucher disease. Other symptoms may include:
-
Bone pain and fractures
Bone pain
Bone pain or tenderness is aching or other discomfort in one or more bones.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Cognitive impairment (decreased thinking ability)
- Easy bruising
-
Enlarged spleen
Enlarged spleen
Splenomegaly is a larger-than-normal spleen. The spleen is an organ in the upper left part of the belly.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
Enlarged liver
Enlarged liver
Enlarged liver refers to swelling of the liver beyond its normal size. Hepatomegaly is another word to describe this problem. If both the liver and ...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
Fatigue
Fatigue
Fatigue is a feeling of weariness, tiredness, or lack of energy.
Read Article Now Book Mark Article - Heart valve problems
- Lung disease (rare)
- Seizures
- Severe swelling at birth
Swelling
Swelling is the enlargement of organs, skin, or other body parts. It is caused by a buildup of fluid in the tissues. The extra fluid can lead to a ...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Skin changes
Exams and Tests
The health care provider will perform a physical exam and ask about the symptoms.
The following tests may be done:
- Blood test to look for enzyme activity
-
Bone marrow aspiration
Bone marrow aspiration
Bone marrow is the soft tissue inside bones that helps form blood cells. It is found in the hollow part of most bones. Bone marrow aspiration is th...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
MRI
MRI
A magnetic resonance imaging (MRI) scan is an imaging test that uses powerful magnets and radio waves to create pictures of the body. It does not us...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
CT
CT
A computed tomography (CT) scan is an imaging method that uses x-rays to create pictures of cross-sections of the body. Related tests include:Abdomin...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
X-ray of the skeleton
X-ray of the skeleton
A skeletal x-ray is an imaging test used to look at your bones. It is used to detect fractures, tumors, or conditions that cause wearing away (degen...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Genetic testing
Treatment
Gaucher disease can't be cured. But treatments can help control and may improve symptoms.
Medicines may be given to:
- Replace the missing GBA (enzyme replacement therapy) to help reduce spleen size, bone pain, and improve thrombocytopenia.
- Limit production of fatty chemicals that build up in the body.
Other treatments include:
- Medicines for pain
- Surgery for bone and joint problems, or to remove the spleen
- Blood transfusions
Support Groups
These groups can provide more information on Gaucher disease:
- National Gaucher Foundation -- www.gaucherdisease.org
- MedlinePlus - Gaucher disease.-- medlineplus.gov/genetics/condition/gaucher-disease/
- National Organization for Rare Diseases -- rarediseases.org/rare-diseases/gaucher-disease rarediseases.org/rare-diseases/gaucher-disease
Outlook (Prognosis)
How well a person does depends on their subtype of the disease. The infantile form of Gaucher disease (Type 2) may lead to early death. Most affected children die before age 5.
Adults with the type 1 form of Gaucher disease can expect normal life expectancy with enzyme replacement therapy.
Possible Complications
Complications of Gaucher disease may include:
-
Seizures
Seizures
A seizure is the physical changes in behavior that occurs during an episode of specific types of abnormal electrical activity in the brain. The term ...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
Anemia
Anemia
Anemia is a condition in which the body does not have enough healthy red blood cells. Red blood cells provide oxygen to body tissues. Different type...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article -
Thrombocytopenia
Thrombocytopenia
Thrombocytopenia is any disorder in which there is an abnormally low amount of platelets. Platelets are parts of the blood that help blood to clot. ...
Read Article Now Book Mark Article - Bone problems
Prevention
Genetic counseling is recommended for prospective parents with a family history of Gaucher disease. Testing can determine if parents carry the gene that could pass on the Gaucher disease. A prenatal test can also tell if a baby in the womb has Gaucher syndrome.
References
Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Defects in metabolism of lipids. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 104.
Krasnewich DM, Sidransky E. Lysosomal storage diseases. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 26th ed. Philadelphia, PA: Elsevier; 2020:chap 197.
Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18.
-
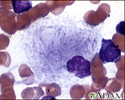
Gaucher cell - photomicrograph - illustration
Gaucher disease is called a lipid storage disease where abnormal amounts of lipids called glycosphingolipids are stored in special cells called reticuloendothelial cells. Classically, the nucleus is pushed off to the side and the remainder of the cell is filled with abnormal lipids.
Gaucher cell - photomicrograph
illustration
-
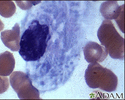
Gaucher cell - photomicrograph #2 - illustration
Gaucher disease is called a lipid storage disease where abnormal amounts of lipids called glycosphingolipids are stored in special cells called reticuloendothelial cells. Classically, the nucleus is pushed off to the side and the remainder of the cell is filled with abnormal lipids.
Gaucher cell - photomicrograph #2
illustration
-
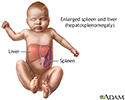
Hepatosplenomegaly - illustration
Liver and spleen enlargement (hepatosplenomegaly) can occur as the result of an inherited disorder in which the liver cannot process glucocerebroside. The buildup of this substance in body tissues can cause severe damage to the central nervous system in infants.
Hepatosplenomegaly
illustration
-
Gaucher cell - photomicrograph - illustration
Gaucher disease is called a lipid storage disease where abnormal amounts of lipids called glycosphingolipids are stored in special cells called reticuloendothelial cells. Classically, the nucleus is pushed off to the side and the remainder of the cell is filled with abnormal lipids.
Gaucher cell - photomicrograph
illustration
-
Gaucher cell - photomicrograph #2 - illustration
Gaucher disease is called a lipid storage disease where abnormal amounts of lipids called glycosphingolipids are stored in special cells called reticuloendothelial cells. Classically, the nucleus is pushed off to the side and the remainder of the cell is filled with abnormal lipids.
Gaucher cell - photomicrograph #2
illustration
-
Hepatosplenomegaly - illustration
Liver and spleen enlargement (hepatosplenomegaly) can occur as the result of an inherited disorder in which the liver cannot process glucocerebroside. The buildup of this substance in body tissues can cause severe damage to the central nervous system in infants.
Hepatosplenomegaly
illustration
Review Date: 10/13/2022
Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 All rights reserved.
All rights reserved.