Cystic fibrosis
Cystic fibrosis is a disease that causes thick, sticky mucus to build up in the lungs, digestive tract, and other areas of the body. It is one of the most common chronic lung diseases in children and young adults. It is a life-threatening disorder.
Cystic fibrosis - Animation
Parents can pass all kinds of different traits to their children, from blue eyes to blonde hair. Sometimes, parents can also pass the genes for certain diseases to their kids. Cystic fibrosis is one very serious inherited disease that makes it hard for children to breathe and digest food. Let's talk about cystic fibrosis. Genes are the coded instructions that tell our bodies how to operate. Usually, the code is correct and everything runs smoothly. But sometimes, the code is incorrect because of a defective gene or genes. In the case of cystic fibrosis, a faulty gene causes the body to produce an abnormally thick, sticky fluid called mucus. This mucus clogs the lungs, making it hard to breathe. It also gets stuck in the pancreas, making it harder for the body to break down and digest food. Millions of Americans carry the cystic fibrosis, or CF gene. Fortunately, most of them don't have cystic fibrosis. That's because you need to inherit one faulty gene from each parent to actually get the disease. Kids who are born with cystic fibrosis start showing signs very early. Newborns don't grow or gain weight as quickly as they should, and they don't make bowel movements because their bodies aren't digesting food properly. As these children get older, they may have symptoms like coughing and fatigue from the mucus in their lungs, and nausea and stomach pain from the mucus in their pancreas. They'll also get pneumonia and other lung infections more often than normal. So, you may now be asking, how do you know if your child has cystic fibrosis? Doctors diagnose cystic fibrosis using a blood test that looks for the CF gene. There is also a sweat test, which looks for saltier-than-normal sweat, another symptom of cystic fibrosis. Doctors may use other tests, such as a chest x-ray or upper GI series, to check for lung and bowel problems caused by CF. It's a good idea to get treated at a center that specializes in cystic fibrosis because they're up on all the latest therapies. For lung problems, treatments include inhaled medicines to open the airways, medicine to thin mucus and make it easier to cough up, and antibiotics to prevent lung infections. Some people may eventually need a lung transplant. For bowel problems, you'll need to eat a special diet that's higher in protein and calories to make up for the nutrients you're losing. You may also take vitamin supplements. The outlook for people with cystic fibrosis is better today than ever before. Thanks to new treatments, people with this disease can live well into adulthood. They can go to school, play sports, and get a job like everyone else. But because the symptoms of cystic fibrosis are so serious, it's very important to stay on top of treatment, and to call the doctor right away if symptoms get worse. Anyone with a family history of the disease may want to get screened for the CF gene before they decide to have children.
Causes
Cystic fibrosis (CF) is a disease that is passed down through families. It is caused by a defective gene that makes the body produce abnormally thick and sticky fluid, called mucus. This mucus builds up in the breathing passages of the lungs and in the pancreas.
The buildup of mucus results in life-threatening lung infections and serious digestion problems. The disease may also affect the sweat glands and a man's reproductive system.
Many people carry a CF gene, but do not have symptoms. This is because for a person to get CF, they must inherit 2 defective genes, 1 from each parent. Some Americans have the CF gene. It is more common among those of northern or central European descent.
Most children with CF are diagnosed by age 2, especially as newborn screening is performed across the United States. For a small number, the disease is not detected until age 18 or older. These children often have a milder form of the disease.
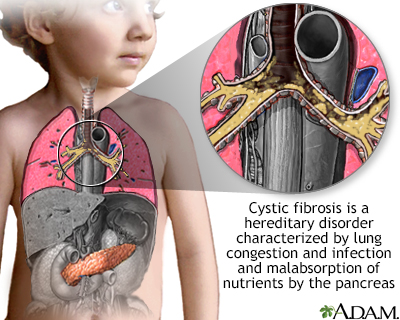
Cystic fibrosis
Cystic fibrosis is the most common cause of chronic lung disease in children and young adults, and the most common fatal hereditary disorder affecting white people in the United States.
Symptoms
Symptoms in newborns may include:
- Delayed growth
Delayed growth
Delayed growth is poor or abnormally slow height or weight gains in a child younger than age 5. This may just be normal, and the child may outgrow i...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Failure to gain weight normally during childhood
- No bowel movements in first 24 to 48 hours of life
- Salty-tasting skin
Symptoms related to bowel function may include:
- Belly pain from severe constipation
- Increased gas, bloating, or a belly that appears swollen (distended)
- Nausea and loss of appetite
- Stools that are pale or clay-colored, foul smelling, have mucus, or that float
Stools that are pale or clay-colored
Stools that are pale, clay, or putty-colored may be due to problems in the biliary system. The biliary system is the drainage system of the gallblad...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Weight loss
- History of rectal prolapse
Symptoms related to the lungs and sinuses may include:
- Coughing or increased mucus in the sinuses or lungs
- Fatigue
Fatigue
Fatigue is a feeling of weariness, tiredness, or lack of energy.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Nasal congestion caused by nasal polyps
- Repeated episodes of pneumonia (symptoms of pneumonia in someone with cystic fibrosis include fever, increased coughing and shortness of breath, increased mucus, and loss of appetite)
Pneumonia
Pneumonia is inflamed or swollen lung tissue due to infection with a germ. This article covers community-acquired pneumonia (CAP). This type of pneu...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Sinus pain or pressure caused by infection or polyps
Polyps
Nasal polyps are soft, sac-like growths on the lining of the nose or sinuses.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article
What happens when a person has cystic fibrosis (CF)? - Animation
Accumulation of mucus contributes to airflow obstruction and predisposes the patient to chronic respiratory infection, which can in turn lead to bronchiectasis and numerous other complications throughout the body. Because CF is caused by an inborn mutation, symptoms of disease typically manifest in childhood. Symptoms of CF include: chronic sinusitis, nasal obstruction, nasal polyps, and cough. Over time, persistent cough produces thick, greenish sputum that may also contain pus. Survival has improved with new therapeutic techniques, and patient life expectancy has increased from late childhood to approximately 40 years.
Symptoms that may be noticed later in life:
- Infertility (in men)
- Repeated inflammation of the pancreas (pancreatitis)
Pancreatitis
Acute pancreatitis is sudden swelling and inflammation of the pancreas.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Respiratory symptoms
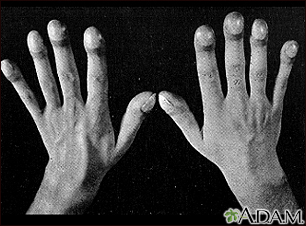
- Clubbed fingers
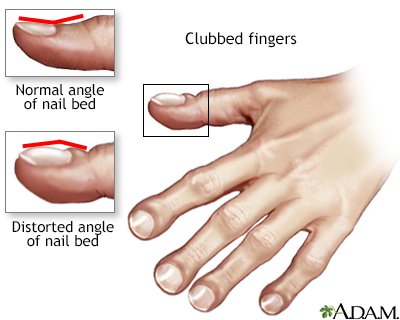
Clubbed fingers
Clubbed fingers is a symptom of disease, often of the heart or lungs which cause chronically low blood levels of oxygen. Diseases which cause malabsorption, such as cystic fibrosis or celiac disease can also cause clubbing.
Exams and Tests
A blood test is done to help detect CF. The test looks for changes in the CF gene. Other tests used to diagnose CF include:
- Immunoreactive trypsinogen (IRT) test is a standard newborn screening test for CF. A high level of IRT suggests possible CF and requires further testing.
- Sweat chloride test is the standard diagnostic test for CF. A high salt level in the person's sweat is a sign of the disease.
Sweat chloride test
Sweat electrolytes is a test that measures the level of chloride in sweat. Sweat chloride test is the standard test used to diagnose cystic fibrosis...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article
Other tests that identify problems that can be related to CF include:
- Chest x-ray or CT scan
Chest x-ray
A chest x-ray is an x-ray of the chest, lungs, heart, large arteries, ribs, and diaphragm.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark ArticleCT scan
A computed tomography (CT) scan is an imaging method that uses x-rays to create pictures of cross-sections of the body. Related tests include:Abdomin...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Fecal fat test
Fecal fat test
The fecal fat test measures the amount of fat in the stool. This can help gauge the percentage of dietary fat that the body does not absorb....
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Lung function tests
Lung function tests
Pulmonary function tests are a group of tests that measure breathing and how well the lungs are functioning.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Measurement of pancreatic function (stool pancreatic elastase)
- Secretin stimulation test
Secretin stimulation test
The secretin stimulation test measures the ability of the pancreas to respond to a hormone called secretin. The small intestine produces secretin wh...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Trypsin and chymotrypsin in stool
Trypsin and chymotrypsin in stool
Trypsin and chymotrypsin are substances released from the pancreas during normal digestion. When the pancreas does not produce enough trypsin and ch...
ImageRead Article Now Book Mark Article - Upper GI and small bowel series
Upper GI and small bowel series
An upper GI and small bowel series is a set of x-rays taken to examine the esophagus, stomach, and small intestine. Barium enema is a different test ...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Lung cultures (obtained by sputum, bronchoscopy or throat swab)
Treatment
An early diagnosis of CF and treatment plan can improve both survival and quality of life. Follow-up and monitoring are very important. When possible, care should be received at a cystic fibrosis specialty clinic. When children reach adulthood, they should transfer to a cystic fibrosis specialty center for adults.
Treatment for lung problems includes:
- Antibiotics to prevent and treat lung and sinus infections. They may be taken by mouth, or given in the veins or by breathing treatments. People with CF may take antibiotics only when needed, or all the time. Doses are often higher than normal.
- Inhaled medicines to help open the airways.
- Other medicines that are given by a breathing treatment to thin mucus and make it easier to cough up are DNAse enzyme therapy and highly concentrated salt solutions (hypertonic saline).
- Flu vaccine and pneumococcal conjugate and polysaccharide vaccines (ask your health care provider for the best age to get these).
Vaccines
Vaccines are used to boost your immune system, reduce the risk of infection, and lessen the severity of infections, including those that cause seriou...
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Lung transplant is an option in some cases.
Lung transplant
Lung transplant is surgery to replace one or both diseased lungs with healthy lungs from a human donor.
 ImageRead Article Now Book Mark Article
ImageRead Article Now Book Mark Article - Oxygen therapy may be needed as lung disease gets worse.
Lung problems are also treated with therapies to thin the mucus. This makes it easier to cough the mucus out of the lungs.
These methods include:
- Activity or exercise that causes you to breathe deeply
- Devices that are used during the day to help clear the airways of too much mucus
- Manual chest percussion (or chest physiotherapy), in which a family member or a therapist lightly claps the person's chest, back, and area under the arms
Treatment for bowel and nutritional problems may include:
Nutritional problems
Cystic fibrosis (CF) is a life-threatening disease that causes thick, sticky mucus to build up in the lungs and digestive tract. People with CF need...
Read Article Now Book Mark Article- A special diet high in protein and calories for older children and adults
- Pancreatic enzymes to help absorb fats and protein, which are taken with every meal
- Vitamin supplements, especially vitamins A, D, E, and K
- Your provider can advise other treatments if you have very hard stools
- Medicines to treat diabetes caused by CF
Ivacaftor, lumacaftor, tezacaftor, and elexacaftor are medicines that treat certain types of CF.
- They improve the function of one of the defective genes that causes CF.
- Up to 90% of patients with CF and eligible for one or more of these medicines alone or in combination.
- As a result, there is less buildup of thick mucus in the lungs. Other CF symptoms are improved as well.
Care and monitoring at home should include:
- Avoiding smoke, dust, dirt, fumes, household chemicals, fireplace smoke, and mold or mildew.
- Giving plenty of fluids, especially to infants and children in hot weather, when there is diarrhea or loose stools, or during extra physical activity.
- Exercising 2 or 3 times each week. Swimming, jogging, and cycling are good options.
- Clearing or bringing up mucus or secretions from the airways. This must be done 1 to 4 times each day. Patients, families, and caregivers must learn about doing chest percussion and postural drainage to help keep the airways clear.
- No contact with other people with CF is recommended as they can exchange infections (does not apply to family members).
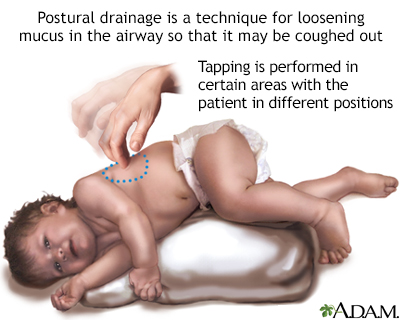
Postural drainage
There are 6 to 12 positions a person with pulmonary disease may take to drain mucus from a certain part of the lungs. Another person may tap in certain areas to help loosen the mucus and allow it to be coughed out. Other ways to relieve the lung congestion of cystic fibrosis or bronchiectasis include percussion vests and inhaled aerosols.
Support Groups
You can ease the stress of illness by joining a cystic fibrosis support group. Sharing with others who have common experiences and problems can help your family to not feel alone.
Cystic fibrosis support group
The following organizations are good resources for information on cystic fibrosis:Centers for Disease Control and Prevention -- www. cdc. gov/cystic-...
Read Article Now Book Mark ArticleOutlook (Prognosis)
Most children with CF stay in good health until they reach adulthood. They are able to take part in most activities and attend school. Many young adults with CF finish college or find jobs.
Lung disease eventually worsens to the point where the person is disabled. Today, the average life span for people with CF who live to adulthood is about 44 years.
Death is most often caused by lung complications.
Possible Complications
The most common complication is chronic respiratory infection.
Other complications include:
- Bowel problems, such as gallstones, intestinal blockage, and rectal prolapse
- Coughing up blood
- Chronic respiratory failure
- Diabetes
- Infertility
- Liver disease or liver failure, pancreatitis, biliary cirrhosis
- Malnutrition
- Nasal polyps and sinusitis
- Osteoporosis and arthritis
- Pneumonia that keeps coming back
- Pneumothorax
- Right-sided heart failure (cor pulmonale)
- Colorectal cancer
When to Contact a Medical Professional
Contact your provider if an infant or child has symptoms of CF, and experiences:
- Fever, increased coughing, changes in sputum or blood in sputum, loss of appetite, or other signs of pneumonia
- Increased weight loss
- More frequent bowel movements or stools that are foul-smelling or have more mucus
- Swollen belly or increased bloating
Contact your provider if a person with CF develops new symptoms or if symptoms get worse, particularly severe breathing difficulty or coughing up blood.
Breathing difficulty
Breathing difficulty may involve:Difficult breathing Uncomfortable breathingFeeling like you are not getting enough air

Prevention
CF cannot be prevented. Screening those with a family history of the disease may detect the CF gene in many carriers.
Reviewed By
Charles I. Schwartz, MD, FAAP, Clinical Assistant Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, General Pediatrician at PennCare for Kids, Phoenixville, PA. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
Eagan ME, Schechter MS, Voynow JA. Cystic fibrosis. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 432.
Grasemann H. Cystic fibrosis. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 77.
Solomon GM, Hoover W, Sorscher EJ, Rowe SM. Cystic fibrosis: diagnosis and management. In: Broaddus VC, Ernst JD, King TE, et al, eds. Murray and Nadel's Textbook of Respiratory Medicine. 7th ed. Philadelphia, PA: Elsevier; 2022:chap 68.
Phan H, Daines CL. Cystic fibrosis. In: Kellerman RD, Rakel DP, Heidelbaugh JJ, Lee EM, eds. Conn's Current Therapy 2024. Philadelphia, PA: Elsevier; 2024:923-928.


 All rights reserved.
All rights reserved.